
Antoine DELINIÈRE
In vitro and in vivo functional characterization of KCNH2 missense variants associated with congenital long-QT syndrome type 2
Summary
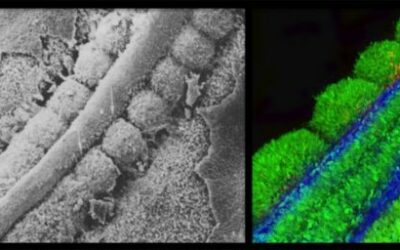
Le gène KCNH2 code pour la sous-unité α du canal hERG (KV11.1) qui joue un rôle majeur dans la repolarisation des cardiomyocytes ventriculaires. Les variations de KCNH2 entraînant une perte de fonction de hERG sont responsables du syndrome du QT long congénital (LQTS) de type 2 (LQT2), une maladie rythmique héréditaire induisant un risque de mort subite. L’impact de la majorité des variations faux-sens de KCNH2 est cependant inconnu, certaines n’ont aucune conséquence fonctionnelle. Le degré de sévérité de la perte de fonction est par ailleurs variant-dépendant. Enfin, le mécanisme le plus fréquemment responsable de la perte de fonction de hERG dans le contexte d’une variation faux-sens est un défaut de trafic intracellulaire du canal. Ce mécanisme déficient pourrait être potentiellement corrigé par plusieurs molécules candidates. Nous proposons une approche combinée in vitro et in vivo pour identifier les variations faux-sens de KCNH2 induisant une perte de fonction de hERG, quantifier la sévérité de cette perte de fonction et évaluer le trafic intracellulaire du canal. Nous avons identifié une nouvelle variation de KCNH2 (G603S). Par une étude de ségrégation familiale nous avons montré que cette variation était associée à l’état homozygote à un phénotype de LQTS sévère et l’état hétérozygote à un phénotype de LQT2 atténué. Nous avons ensuite réalisé une évaluation fonctionnelle in vitro de hERG-G603S par la technique du two-electrode voltage-clamp sur le système d’expression hétérologue des ovocytes de Xenopus laevis. La conductance macroscopique était significativement réduite lorsque l’ARNc de KCNH2 G603S était injecté seul dans les ovocytes, témoignant d’un effet perte de fonction. La cinétique d’inactivation et la voltage-dépendance de l’activation n’étaient pas impactées par la variation. Ces données ne préjugeaient pas du mécanisme de la perte de fonction, car le trafic intracellulaire de hERG est facilité in vitro sur les ovocytes de xénopes. Nous avons ensuite évalué l’intérêt de Caenorhabditis elegans comme modèle in vivo du LQT2. C. elegans possède en effet un orthologue de hERG, le canal UNC-103 (codé par unc-103). Ce canal est notamment exprimé dans les muscles vulvaires et les jonctions neuromusculaires (JNM). Grâce à la technique d’édition génétique du CRISPR/Cas9 nous avons pu marquer le canal UNC-103 par des protéines fluorescentes et insérer des variations humaines de KCNH2 dans le gène unc-103. Nous avons d’abord étudié 2 variations perte de fonction sévères dont les mécanismes avaient été élucidés dans des modèles in vitro (G604S : défaut de trafic intracellulaire, G628S : défaut de sélectivité) ainsi que le variant G603S, dont le mécanisme restait à découvrir. Nous avons pu quantifier la perte de fonction et évaluer le trafic intracellulaire du canal en combinant des études quantitatives du phénotype de ponte et de la fluorescence à la JNM en microscopie confocale. Les données in vivo de G604S et G628S étaient cohérentes avec les autres modèles (perte de fonction sévère pour les deux variations, trafic intracellulaire altéré pour G604S et préservé pour G628S). G603S entraînait une perte de fonction modérée et une altération significative du trafic intracellulaire du canal. Enfin, nous avons pu quantifier in vivo la perte de fonction induite par 4 autres variations de KCNH2 impliquées dans le LQT2 (A561T, G572S, T613M et F640V). Ainsi, C. elegans est un modèle in vivo simple et puissant pour quantifier l’impact fonctionnel de variants faux-sens de KCNH2 et évaluer leur éventuel effet sur le trafic intracellulaire de hERG. Ce nouveau modèle ouvre de nombreuses perspectives de recherche, comme l’intégration des données fonctionnelles dans l’évaluation du risque de mort subite et l’évaluation in vivo et variant-spécifique de molécules candidates pour le traitement du LQT2.
